Utilizing Maze Compass™, we are harnessing the power of human genetics to develop novel, small molecule precision medicines for patients living with renal, cardiovascular and related metabolic diseases, including obesity.
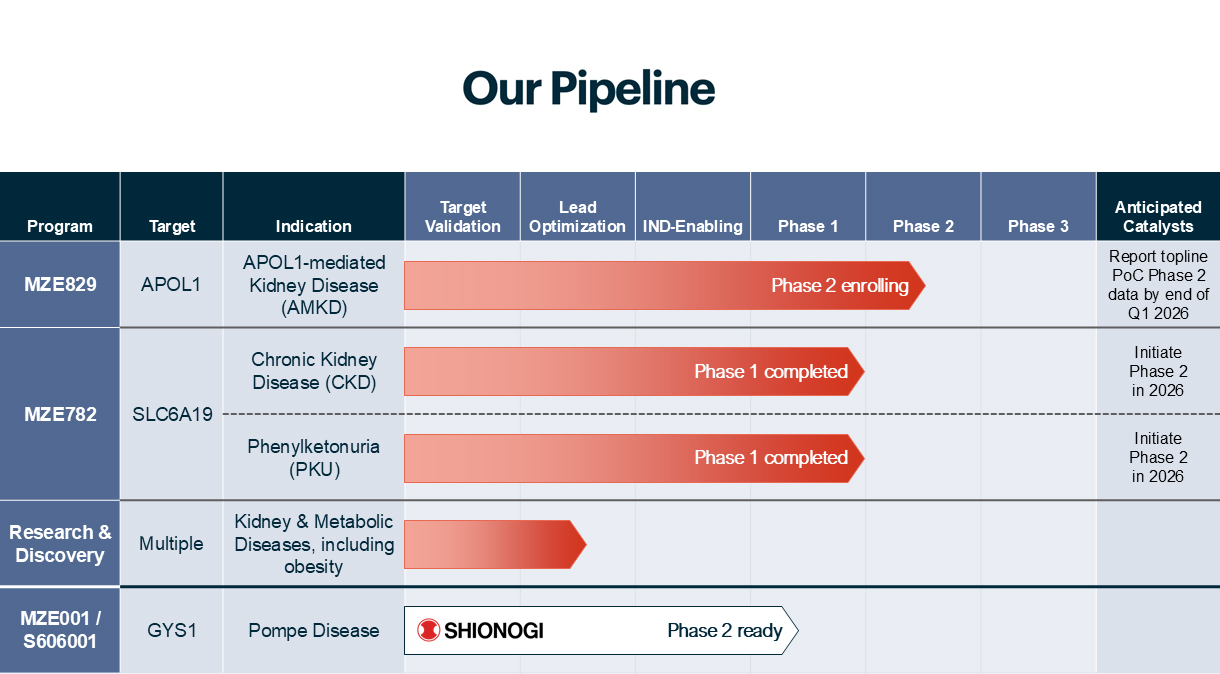
MZE829 for APOL1-Mediated
Kidney Disease (AMKD)
In the United States, approximately six million, or 13%, of African Americans have mutations of both copies of the high-risk APOL1 gene variants, and are at risk for developing AMKD. It is currently estimated that approximately 20%, or over one million, of those individuals have AMKD.
Click here to learn more
Click here to learn more at www.apol1kdcare.com
For more information about our APOL1-Mediated Kidney Disease study, visit clinicaltrial.gov
MZE782 for Chronic Kidney Disease and PKU
Chronic kidney disease (CKD) impacts approximately 37 million, or 1 in 7 individuals in the U.S. alone.2 Current treatments for CKD do not address the underlying cause and instead focus on slowing the progression. Phenylketonuria (PKU) is an inherited metabolic disorder impacting approximately 60,000 patients worldwide. While PKU treatment options exist, their effectiveness is limited for some patients, and adhering to a low-Phe diet over a lifetime can be challenging for many individuals.
Click here to learn more
2. Source: https://www.cdc.gov/kidneydisease/publications-resources/ckd-national-facts.html